
| Ehlers–Danlos syndrom | |
| Latin: syndroma ehlers-danlos | |
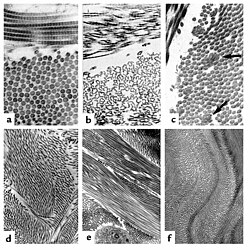 Kollagenet i EDS. (a) Normala fibriller av kollagen har samma storlek och avstånd. Fibriller från en person med dermatosparaxis (b) visar en dramatisk förändring i fibrillernas morfologi vilket ger allvarlig påverkan på bindvävens spänning. Personer med EDS (c) uppvisar fibriller med kompositer. Fibriller från en person med brist på TNX (d) är lika i storlek och det syns inga fibriller med kompositer. TNX-null fibriller (e) är mindre tätt placerade och inte lika väl positionerade till fibrillerna i närheten.
| |
| Klassifikation och externa resurser | |
|---|---|
| ICD-10 | Q79.6 (ILDS Q82.817) |
| ICD-9 | 756.83 |
| Medlineplus | 001468 |
| eMedicine | derm/696 ped/654 |
| MeSH | svensk engelsk |



Ehlers–Danlos syndrom (EDS) är en grupp ärftliga bindvävsavvikelser. EDS orsakas av bristfällig bildning av de normalt starka proteinstrukturer i kroppen som kallas kollagen. Kollagen, ett av kroppens grundläggande byggnadsmaterial, spelar en viktig roll när det gäller att hålla ihop, stärka och ge elasticitet till celler och vävnader. Vid EDS finns en förglesning och förändrad struktur i kollagenkedjorna, som normalt binder ihop sig tre och tre och bildar trådar (fibriller) med stor styrka. Detta gör att hållfastheten i vävnaderna blir sämre.
Skillnaderna mellan de olika typerna är stora. Hypermobilitetstypen för till exempel med sig kronisk smärta, men den drabbade kan nå lika hög ålder som befolkningen i övrigt. Den som har kärltypen har dock inte någon oupphörlig smärta men dör vanligtvis redan i 30-, 40- eller 50-årsåldern av att aorta och/eller andra stora blodkärl brister och orsakar kraftiga inre blödningar.
Symptom
De olika typerna av EDS kan kännas igen av den mix av sjukdomssymptom som dominerar:
- ledöverrörlighet (hypermobilitet), urledvridningar och muskel-, led- och andra smärtor som blir kroniska och ibland mycket svåra
- överelastisk hud och överelasticitet i kroppsliga vävnader och membran (olika typer av väggar hos inre organ)
- alltför bräcklig hud och bristfällig sårläkning
- mycket försvagade blodkärl och svagheter i andra kroppsliga vävnader och membran (till exempel tarmväggar).
De som har EDS får tidigt i livet sin kroniska överansträngningssmärta. Muskelsmärtorna är vanligen knutna till en kronisk trötthet i de muskler som oavbrutet måste stabilisera en led på grund av att de ledband och ligament som egentligen ska sköta stabiliseringen inte gör detta eftersom de är överelastiska. Den överelastiska bindväven innebär att en stor del av musklernas dragningskraft inte överförs till skelettet. Muskelansträngningen vid en rörelse kan därför uppgå till 300 procent av den normala. Nervsmärtor uppstår främst som ett resultat av nötning där nervtrådarna löper genom överrörliga leder eller krampaktigt spända muskler.
Epidemiologi
EDS finns hos alla folk i hela världen, är lika vanlig hos män som hos kvinnor, och förekomsten uppskattas för närvarande till en på 5 000 invånare. En intressant iakttagelse är att de vanligare typerna av EDS förekommer i samma utsträckning även hos folk som i tusentals år levt isolerade från resten av världens befolkning. Exempel är maorierna på Nya Zeeland, aboriginerna i Australien, ursprungsbefolkningen på Nya Guinea samt indianerna i Eldslandet.
Klassifikation
År 2017 utkom en uppdaterad klassifikation av Ehlers–Danlos syndrom som identifierar 13 typer av EDS. Klassifikationen ersätter den från 1998 då ett antal läkare kom överens om diagnoskriterier och nosologi för EDS och angav sex huvudsakliga typer. Klassifikation av EDS har funnits sedan 1988 då 11 typer beskrevs utifrån kliniska tecken och symptom samt nedärvningsmönster.
2017 års klassifikation delar in EDS i 13 sjukdomar:
- Klassisk (cEDS)
- Klassisk-liknande (clEDS)
- Cardiac-valvular (cvEDS)
- Vaskulär (vEDS)
- Hypermobil EDS (hEDS)
- Arthrochalasia (aEDS)
- Dermatosparaxis (dEDS)
- Brittle cornea syndrom (BCS)
- Spondylodysplastic (spEDS)
- Musculocontractural (mcEDS)
- Myopathic (mEDS)
- Periodontal (pEDS)
1998 års klassifikation innefattar följande sex typer av EDS:
- Klassisk typ
Den klassiska typen av EDS var den som först uppmärksammades. Här är det fråga om en sällsynt ärftlig sjukdom som kännetecknas av stor elasticitet i huden, bräcklig hud med en tendens till blödning, dålig sårläkning med bräcklig ärrbildning, och uttalad extensibilitet i lederna ("gummimän"). Huden är sammetslen och mycket töjbar (huden på hakan kan dras upp över munnen och ibland ända upp till nästippen). Huden drabbas mycket lätt av blåmärken. Öronen och ögonlock kan tendera att hänga ned en aning, dislokationer av leder är mycket vanliga, till och med före födseln. Man har senare konstaterat att det finns varierande grad av uttryck mellan drabbade individer, uppenbarligen drabbades inte alla lika svårt.
Läkarvetenskapen gjorde därför en indelning av den klassiska typen i en svårare och en mildare form. Huden är alltifrån något till extremt töjbar. Vissa drabbade kan inte dra ut huden mer än ett par centimeter. Huden är dessutom mer eller mindre skör. Den kan i extremfallet spricka mycket lätt och ha mycket svårt att läka. Ärren blir onormalt tunna och sköra som cigarettpapper. Översträckbarheten i lederna är påfallande och lederna kan lätt gå ur led. Detta gäller särskilt i händer, fingrar, fötter, tår, armbågar, axlar, höfter och knän. Vrickningar, där ledkulorna släpper ur sina lägen, är vanliga.
- Överrörlighetstyp
Hypermobilitetstypen medför överrörlighet i alla leder och därmed luxationer (urledvridningar) och subluxationer (ledglidningar) och felställningar (till exempel i revben). Nästan alla leder kan drabbas, men ofta har individen några leder som drabbas mer än andra: ett knä, en armbåge, ett nyckelben, revben, käken och så vidare. Huden kan vara mjuk och påfallande len, ibland nästan sammetsliknande. Huden är emellertid inte skör och tänjbar som vid den klassiska typen av EDS. Blåmärken förekommer, men variationen mellan olika personer är stor vad gäller omfattning och svårighetsgrad av blåmärken. Personer med denna typ av EDS kan tidigt i livet drabbas av kronisk smärta, som i enskilda fall kan vara oupphörlig och mycket svår på grund av en blandning av olika typer av smärta: muskelsmärtor, ledsmärtor och nervsmärtor. Över 50 procent av de EDS-drabbade har hypermobilitetstypen. Muskelsvaghet kan även förekomma.
- Kärltyp
Kärltypen är den form av EDS som oftast leder till döden i förtid. Knappt 4 procent av dem som har EDS har kärltypen. Det finns en viss överrörlighet i små leder. Huden är mycket tunn, nästan genomskinlig, och kan redan när patienten är ung se åldrad ut. Underhudsblödningar i form av blåmärken uppstår mycket lätt. Det är den uttalade skörheten i artärer, mag/tarmsystem och livmoder som orsakar bristningar och livshotande blödningar.
- Artrochalasi-typ
Artrochalasiatypen har mindre än 1 procent av de EDS-drabbade. Här finns en markant, allomfattande ledöverrörlighet med återkommande luxationer. Ett viktigt kännetecken är att de barn som föds med denna typ av EDS har den ena eller båda höftlederna ur led (höftledsluxation) efter förlossningen. Huden är töjbar och skör, underhudsblödningar uppstår lätt. Muskelsvaghet, skolios och viss benskörhet förekommer.
- Kyfoskolios-typ
Kyfoskoliotisk typ är en mycket sällsynt form av EDS med generell ledöverrörlighet och kraftig muskelsvaghet redan från födseln. Dessutom hör progressiv skolios till denna typ av EDS. Ögonvitan är skör och bristningar förekommer i ögongloben. Huden är skör och bildar atrofiska ärr vid sårläkning. Blåmärken uppstår lätt. Med kyfoskolios-typ kommer en mycket grov trötthet, som en studie visar är mycket ovanlig inom mänskligheten.
- Dermatosparaxis-typ
Dermatosparaxistypen är en ytterst ovanlig form av Ehlers–Danlos syndrom. De flesta komplikationerna har med huden att göra. Huden är mjuk och "degig", lös och överflödig med stora bråck i navel och ljumskar; dessutom är hudskörheten uttalad. Blåmärken uppkommer lätt. Prematurer är vanliga.
Genetik
Ärftligheten vad gäller artrochalasiatypen, hypermobilitetstypen, den klassiska typen och kärltypen är autosomal dominant. Varje nytt barn från en drabbad förälder löper 50 procents risk att ärva EDS.
Två av EDS-typerna — kyfoskoliosis och dermatosparaxis — har ett ärftlighetsmönster som kallas autosomal recessivt. Detta innebär att en kopia av den muterade genen från vardera föräldern måste ärvas för att sjukdomen ska utvecklas. Den som endast ärver en kopia blir "bärare" av defekten utan att uppvisa några symptom. Om denne bärare osannolikt nog skulle hitta en partner med samma form av EDS måste deras barn få den muterade genen från båda föräldrarna för att få sjukdomen.
Diagnos
Om symptomen är måttliga vid EDS leder detta inte sällan till felaktiga diagnoser som exempelvis fibromyalgi och reumatism. Det tar i allmänhet lång tid att få rätt diagnos eller att få någon diagnos alls, eftersom flera landsting saknar specialister på EDS. Eftersom EDS-patienter ska remitteras till olika kliniker beroende på problem – genetiker, hudläkare, reumatolog, ortoped, kirurg – kan det vara svårt att få en läkare som har en helhetsbild.
Namn
Ehlers–Danlos syndrom är namngivet efter två läkare: Edvard Ehlers från Danmark och Henri-Alexandre Danlos från Frankrike. Ehlers–Danlos syndrom kallas även Chernogubovs syndrom, idag uteslutande i rysk litteratur, efter den ryska hudläkaren Nikolaj Alexandrovitj Chernogubov som 1892 beskrev den bindvävsdefekt som numera är känd som Ehlers–Danlos syndrom.