
| Tyrosinemi typ 1 | |
| |
| Klassifikation och externa resurser | |
|---|---|
| ICD-10 | E70.2 |
| ICD-9 | 270.2 |
| OMIM | 276700 |
| DiseasesDB | 13478 |
| eMedicine | ped/2339 |
| MeSH | svensk engelsk |
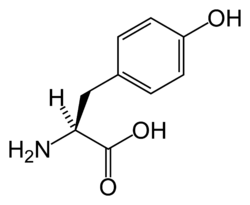
Tyrosinemi typ 1 (TYR1) är den vanligaste och mest allvarliga varianten av tyrosinemi. Det är en medfödd ämnesomsättningssjukdom som är orsakad av att enzymet fumarylacetonacetat hydrolas inte fungerar som det ska. Sjukdomen leder till ansamling av aminosyran tyrosin och dess biprodukter i blodet och ute i vävnaderna eftersom kroppen inte kan metabolisera dessa, och kan utan behandling leda till konsekvenser som hepatomegali.
Genetik
TYR1 drabbar ungefär 1 på 100 000 individer i Sverige, men frekvensen varierar runt om i världen. Sjukdomen är allra vanligast i Québec i Kanada, där den drabbar 1 på 16 000 individer i speciellt i en region av Québec, Saguenay–Lac-Saint-Jean, där 1 på cirka 1800 individer drabbas. TYR1 är en monogen sjukdom med ett autosomalt recessivt ärftlighetsmönster som beror på mutation i genen FAH, position 15q25.1. Genen ansvarar normalt för produktion av ett enzym, fumarylacetoacetat hydrolas (FAH), som spelar en viktig roll i nedbrytning av aminosyran tyrosin. Sjukdomen har kunnat kopplas till ett flertal FAH-mutationer.
Man har lyckats hitta minst 86 mutationer i FAH-genen som man kunnat korrelera med TYR1. Beroende på var i världen sjukdomen dyker upp verkar vissa varianter vara vanligare än andra. IVS12+5G-A är en av de vanligaste FAH-allelerna och det är framförallt den som man funnit hos de drabbade individerna i Québec i Kanada. Mutationen som gett upphov till allelvarianten är en splice site mutation i intron 12, som resulterar i ett onormalt kort enzym som då inte kan fungera korrekt. Detta leder till att tyrosin inte metaboliseras hela vägen och fumarylacetoacetat (FAA) kommer således att ansamlas i levern och njurarna. Förhöjda nivåer av FAA i dessa organ är toxiskt och orsakar framförallt lever- och njurproblem.
FAH-enzymet uttrycks rikligt i framförallt njurar och lever, men finns även i mindre mängder i övriga vävnader i kroppen. FAH är ett av fem enzym som är med i stegen för att bryta ner aminosyran tyrosin, där FAH är ansvarigt för det sista steget i att bryta ner FAA till succinat, fumarat och acetoacetat. Mutation i FAH-genen som orsakar TYR1 leder till att genen producerar ett ostabilt eller inaktivt FAH-enzym. Detta leder till reducerad eller frånvarande enzymaktivitet, vilket innebär att tyrosin inte kan metaboliseras fullständigt. När tyrosin inte bryts ner kommer dess ämnesomsättningsprodukter, bland annat FAA, att ansamlas. FAA är toxiskt och kommer bland annat att ackumuleras i hepatocyter, som orsakar cellulär skada och apoptos. FAA kan även omvandlas till succinylaceton, vilket kan påverka aktiviteten hos två olika enzymer i levern: p-HPPD, så att tyrosinkoncentrationen i levern ökar, samt PBG-syntas som indirekt ökar mängden 5-aminolevulinsyra (δ-ALA).
Symptom
Barn som inte blir behandlade för sjukdomen får en rad varierande symptom, exempelvis hepatomegali, dålig tillväxt, ascites, feber, diarré, kräkningar, leversvikt, gulsot, nedsatt njurfunktion, rakit, och levertumörer. TYR1 förekommer i akut och kronisk form. Barn med akut form brukar insjukna redan under de första månaderna och utan behandling kan sjukdomen snabbt komma att bli livshotande på grund av leversvikt och koagulationsrubbningar. Om individen istället har den kroniska varianten kommer symptomen inte att visa sig lika snabbt, det kan ske från allt till 1 års ålder, upp till skolåldern. Obehandlad kronisk form leder ofta till för tidig död i och med att det utvecklas elakartade levertumörer. Barn som får tillgång till rätt behandling har dock få eller inga symptom.
Ärftlighet
TYR1 har ett autosomalt recessivt nedärvningsmönster, vilket innebär att en sjuk individ har sjukdomsassocierade varianter av FAH-genen på båda allelerna. Två individer som får barn kan därför båda vara friska bärare av sjukdomen, men ändå kunna föra vidare varsin sjukdomsallel av till barnet som då kommer få dubbel uppsättning av sjukdomsalleler och utveckla sjukdomen. Av eventuella avkommor till två friska bärare är det 25% risk att barnet får dubbel uppsättning av sjukdomsalleler, 50% risk att barnet blir bärare av en sjukdomsallel och 25% chans att barnet får dubbel uppsättning friska alleler.
PKU-test
PKU-test görs i Sverige och många andra länder på nyfödda barn för att hitta en rad ovanliga, medfödda sjukdomar. En av dessa sjukdomar är TYR1. Provet går till genom att man screenar nyfödda barn med hjälp av tandem-masspektrometri för att detektera metaboliter i urin- och blodprov. Det är effektivare att undersöka metaboliterna eftersom dessa medfödda sjukdomar ofta kan orsakas av ett flertal mutationer.
Typiska biokemiska observationer man kan se hos individer med just TYR1 inkluderar: ökad koncentration succinylaceton i blodet och urinen; förhöjd koncentration av tyrosin i blodet; ökad koncentration av tyrosinmetaboliter i urinen; ökad halt δ-ALA i urinen; allmän negativ påverkan på njurar och lever. Om PKU-provet visar sig vara positivt görs ofta en molekylärgenetisk analys för de vanligaste patogena varianterna av FAH-genen.
Behandling
Behandlingen för TYR1 är livslång och sker med dioxygenashämmare som blockerar nedbrytningen av tyrosin i det andra steget så att de giftiga biprodukterna inte kan ansamlas. Men då tyrosinnivån i blodet stiger så kombineras behandlingen med en proteinfattig kost tillsammans med aminosyratillskott fria från tyrosin och fenylalanin, för att få i sig så lite av dem som möjligt.